
La fibrose kystique (FK ; OMIM #219700), ou mucoviscidose, est une maladie génétique autosomique récessive qui affecte différents organes. Les patients atteints de FK souffrent généralement d’une maladie pulmonaire obstructive accompagnée d’une infection bactérienne chronique, d’une insuffisance enzymatique pancréatique et d’un excès de sel dans la sueur. La FK est causée par des mutations du gène CFTR (OMIM #602421) qui code pour la protéine CFTR. Ce canal à anions régule le transport de l’eau et des ions et entretient l’hydratation de la surface épithéliale.
La FK présente un mode de transmission autosomique récessive typique. Bien que la délétion F508 (F508del ou DF508) représente environ 90 % de tous les cas de FK, plus de 2100 variants du gène CFTRont été identifiés. La répartition et la fréquence des variants varient selon les différents groupes ethniques et régions. En Europe, 82,4 % des patients présentent au moins une mutation F508del, bien que la fréquence soit plus élevée dans le nord de l’Europe que dans le sud.
La mutation F508del de type II est non seulement la mutation la plus fréquente, mais elle provoque également un phénotype de FK plus sévère, car elle diminue l’expression de la membrane du canal et altère également sa fonction.
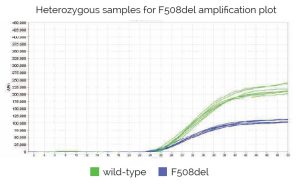
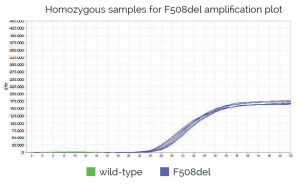
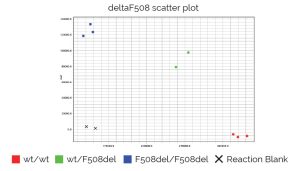
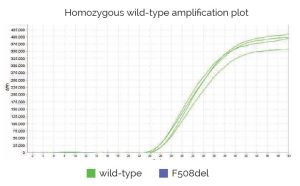
Genvinset® deltaF508 es un kit de diagnostic in vitro semi-automatique pour la détermination qualitative des allèles wt et/ou F508del (dbSNP: rs113993960 NM_000492.4:c.1521_1523delCTT) du gène CFTR (OMIM : 601806) associés à la fibrose kystique, sur des échantillons d’ADN génomique extrait de sang total, à l’aide de la technologie de PCR en temps réel avec sondes TaqMan®.
Le patient adressé par le spécialiste de la santé concerné (par exemple, pneumologue), compte tenu de la compatibilité des symptômes présentés (infections pulmonaires ou pneumonie, respiration sifflante ou rauque, toux avec mucus épais, mouvements de ventre volumineux et gras, constipation ou diarrhée, difficulté à prendre du poids ou à grandir, sueur très salée, et/ou antécédents familiaux, par exemple un ascendant direct avec diagnostic de fibrose kystique) peut être soumis à un test de détermination de la mutation F508del dans le gène CFTR. Ce test peut également être destiné à un patient nouveau-né chez qui la confirmation moléculaire de la fibrose kystique est effectuée après avoir observé des écarts par rapport au test biochimique de dépistage néonatal, sur la base d’une valeur de trypsine immunoréactive (IRT) anormale. Les résultats de ce test ne doivent pas constituer la seule base des décisions thérapeutiques et doivent être utilisés comme une aide au diagnostic, en association avec les résultats d’autres marqueurs de la maladie.
L’utilisateur prévu du kit est un technicien formé et qualifié pour réaliser le protocole conformément aux instructions d’utilisation et procéder à l’interprétation des résultats.